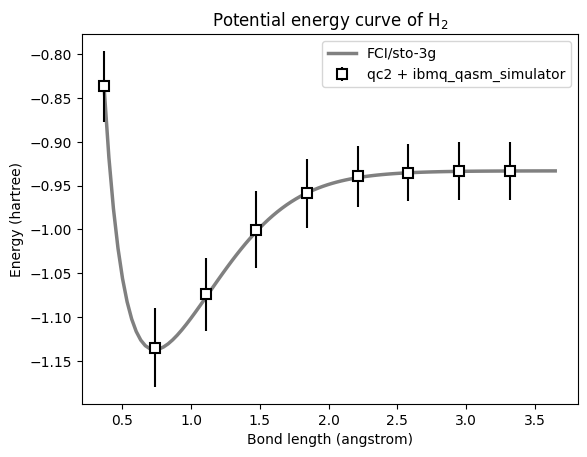
Example 4 (Advanced options): PEC of H\(_{2}\) using Qiskit IBM Runtime Service.
In this section, we demonstrate how to use qc2 alongside Qiskit and the IBM Quantum Runtime Service for calculating the potential energy curve of the hydrogen molecule. We utilize the EstimatorV2 primitive as implemented in the qiskit-ibm-runtime package, in conjunction with the ibmq_qasm_simulator, to simulate a real quantum backend.
Import required packages
[1]:
import numpy as np
# ASE Atoms object
from ase import Atoms
# IBMQ runtime service packages
from qiskit_ibm_runtime import (
QiskitRuntimeService,
EstimatorV2 as Estimator,
EstimatorOptions,
Session
)
# Qiskit-related packages
from qiskit_nature.second_q.circuit.library import HartreeFock, UCCSD
from qiskit_nature.second_q.mappers import JordanWignerMapper
from qiskit_algorithms.optimizers import COBYLA
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
# ignore package import warnings
import warnings
warnings.filterwarnings('ignore')
# qc2 packages
from qc2.data import qc2Data
from qc2.ase import PySCF
Initiate IBM runtime service
In the following steps, we assume that you have already created an IBM Quantum account and saved your accound credentials (via your personal token) on disk; for further info click here.
[2]:
# instantiating `QiskitRuntimeService`
service = QiskitRuntimeService()
# run on simulator
backend = service.backend("ibmq_qasm_simulator")
# Use the following line if you want to run on a real quantum backend
# backend = service.least_busy(simulator=False)
options = EstimatorOptions()
# set seed for reproducible runs
options.seed_estimator = 42
# error supression options
options.optimization_level = 1
# error mitigation options
options.resilience_level = 0 # 2 = ZNE
# execution options
options.default_shots = 6000
# instantiate the Estimator to calculate expectation values
estimator = Estimator(backend=backend, options=options)
Set up calculation details
[3]:
# define activate space
n_active_electrons = (1, 1)
n_active_spatial_orbitals = 2
# define the type of fermionic-to-qubit transformation
mapper = JordanWignerMapper()
# define file to write the calculated data to
output_file = 'h2_pec_ibmq_qasm_simulator.data'
file = open(output_file, "w", encoding='utf-8')
# set up equilibrium geometry and initial circuit parameters in rads
req = 0.737166
req_params = [1.57079357, 1.57087253, 1.45852109]
# define vector of internuclear distances to loop over
r_distances = [r*req for r in np.arange(0.5, 5, 0.5)]
Run VQE for each selected geometry
[4]:
for r in r_distances:
# set Atoms object
mol = Atoms(
'H2',
positions=[[0., 0., 0.],
[0., 0., r]]
)
# instantiate qc2Data class
qc2data = qc2Data(
molecule=mol,
filename='h2.hdf5',
schema='qcschema'
)
# specify the qchem calculator
qc2data.molecule.calc = PySCF(method='scf.RHF', basis='sto-3g')
# run calculation and save qchem data in the hdf5 file
qc2data.run()
# set up qubit Hamiltonian and core energy based on given activate space
e_core, qubit_op = qc2data.get_qubit_hamiltonian(
n_active_electrons,
n_active_spatial_orbitals,
mapper,
format='qiskit'
)
# build up the initial quantum circuit
reference_state = HartreeFock(
n_active_spatial_orbitals,
n_active_electrons,
mapper,
)
# set up the ansatz using unitary CC as variational form
ansatz = UCCSD(
n_active_spatial_orbitals,
n_active_electrons,
mapper,
initial_state=reference_state,
)
# create initial callback dictionary
callback_dict = {
"backend": None,
"iters": 0,
"cost_history": [],
"parameters": [req_params],
"gs_energy_history": [],
"variance": []
}
def cost_func(params: list) -> float:
"""Return electronic energy from Estimator for given set of parameters.
Args:
params: list
List of VQE parameters.
Returns:
energy: float
Calculated electronic ground-state energy.
"""
# perform transpilation
pm = generate_preset_pass_manager(backend=backend, optimization_level=0)
isa_circuit = pm.run(ansatz)
isa_observable = qubit_op.apply_layout(isa_circuit.layout)
# run job
job = estimator.run(
[(isa_circuit, isa_observable, params)]
)
result = job.result()
energy = result[0].data.evs
variance = result[0].data.stds
parameters = list(params)
callback_dict["iters"] += 1
callback_dict["cost_history"].append(energy)
callback_dict["backend"] = job.backend()
callback_dict["gs_energy_history"].append(energy+e_core)
callback_dict["variance"].append(variance)
callback_dict["parameters"].append(parameters)
# print intermediate data if needed
# print("=== COST FUNCTION SUMMARY ===")
# print(f">>> Job ID: {job.job_id()}")
# print(f">>> Job Status: {job.status()}")
# print("=============================")
# print(f">>> Job Input: {job.inputs}")
# print("=============================")
# print(f">>> Backend: {job.backend()}")
# print(f">>> {result}")
# print("=============================")
# print(f">>> Expectation value (Hartree): {energy}")
# print(f">>> Total ground state energy (Hartree): {energy+e_core}")
# print("=============================\n")
return energy
# initiate Qiskit Runtime Section
with Session(service=service, backend=backend) as session:
# initial circuit parameters
initial_theta = np.array(callback_dict['parameters'][-1])
# set up the classical optimizer
optimizer = COBYLA(maxiter=300)
# find the best circuit parameters that minimizes the electronic energy
res = optimizer.minimize(
cost_func,
x0=initial_theta
)
print(f">>> Final ground state energy (hartree): {res.fun+e_core}\n")
# write relevant data to file
data_to_write = f"{r} {callback_dict['gs_energy_history'][-1]} " \
f"{callback_dict['variance'][-1]}\n"
file.write(data_to_write)
# use the optimized circuit parameters as guess for the next geometry
req_params = callback_dict['parameters'][-1]
session.close()
file.close()
* Reference energy (Hartree): -0.8323959925858835
* Saving qchem data in h2.hdf5
>>> Final ground state energy (hartree): -0.8366257350658743
* Reference energy (Hartree): -1.11690055771897
* Saving qchem data in h2.hdf5
>>> Final ground state energy (hartree): -1.1349489643349955
* Reference energy (Hartree): -1.0347677703871998
* Saving qchem data in h2.hdf5
>>> Final ground state energy (hartree): -1.0739508097501043
* Reference energy (Hartree): -0.9186044326889025
* Saving qchem data in h2.hdf5
>>> Final ground state energy (hartree): -1.0002826958494946
* Reference energy (Hartree): -0.8185133354304801
* Saving qchem data in h2.hdf5
>>> Final ground state energy (hartree): -0.9588193874386988
* Reference energy (Hartree): -0.7444704693706197
* Saving qchem data in h2.hdf5
>>> Final ground state energy (hartree): -0.939401703419809
* Reference energy (Hartree): -0.6935395490285441
* Saving qchem data in h2.hdf5
>>> Final ground state energy (hartree): -0.9350495720648158
* Reference energy (Hartree): -0.6597173498273768
* Saving qchem data in h2.hdf5
>>> Final ground state energy (hartree): -0.9335246431974368
* Reference energy (Hartree): -0.6376873315318315
* Saving qchem data in h2.hdf5
>>> Final ground state energy (hartree): -0.9329587957364209
Calculate PEC using PySCF for comparison
With our VQE points done, let’s calculate the PEC using a classical qchem backend to compare with, but now using a finer grid of internuclear distances.
[5]:
import numpy as np
import pyscf
from pyscf import gto, scf, fci
# set up equilibrium geometry and vector of internuclear distances
req = 0.737166
r_distances = [r*req for r in np.arange(0.5, 5, 0.05)]
# define file to write data to
output_file = 'pyscf_fci_sto3g_h2.data'
with open(output_file, "w", encoding='utf-8') as file:
for r in r_distances:
# run HF
mol_h2 = gto.M(atom=f'H 0 0 0; H 0 0 {r}', basis='sto-3g', verbose=0)
scf_h2 = scf.RHF(mol_h2)
hf_energy = scf_h2.kernel()
# run FCI
hf_h2 = mol_h2.RHF().run()
fci_energy = fci.FCI(hf_h2).kernel()[0]
print(f'* Energy (hartree) {fci_energy:.6f} :: r (angstrom) {r:.3f}')
# write data to file
data_to_write = f"{r} {hf_energy} {fci_energy}\n"
file.write(data_to_write)
* Energy (hartree) -0.841556 :: r (angstrom) 0.369
* Energy (hartree) -0.924999 :: r (angstrom) 0.405
* Energy (hartree) -0.987489 :: r (angstrom) 0.442
* Energy (hartree) -1.034243 :: r (angstrom) 0.479
* Energy (hartree) -1.068956 :: r (angstrom) 0.516
* Energy (hartree) -1.094322 :: r (angstrom) 0.553
* Energy (hartree) -1.112352 :: r (angstrom) 0.590
* Energy (hartree) -1.124586 :: r (angstrom) 0.627
* Energy (hartree) -1.132223 :: r (angstrom) 0.663
* Energy (hartree) -1.136210 :: r (angstrom) 0.700
* Energy (hartree) -1.137302 :: r (angstrom) 0.737
* Energy (hartree) -1.136103 :: r (angstrom) 0.774
* Energy (hartree) -1.133097 :: r (angstrom) 0.811
* Energy (hartree) -1.128672 :: r (angstrom) 0.848
* Energy (hartree) -1.123139 :: r (angstrom) 0.885
* Energy (hartree) -1.116748 :: r (angstrom) 0.921
* Energy (hartree) -1.109701 :: r (angstrom) 0.958
* Energy (hartree) -1.102166 :: r (angstrom) 0.995
* Energy (hartree) -1.094279 :: r (angstrom) 1.032
* Energy (hartree) -1.086157 :: r (angstrom) 1.069
* Energy (hartree) -1.077900 :: r (angstrom) 1.106
* Energy (hartree) -1.069594 :: r (angstrom) 1.143
* Energy (hartree) -1.061316 :: r (angstrom) 1.179
* Energy (hartree) -1.053131 :: r (angstrom) 1.216
* Energy (hartree) -1.045097 :: r (angstrom) 1.253
* Energy (hartree) -1.037263 :: r (angstrom) 1.290
* Energy (hartree) -1.029672 :: r (angstrom) 1.327
* Energy (hartree) -1.022360 :: r (angstrom) 1.364
* Energy (hartree) -1.015354 :: r (angstrom) 1.401
* Energy (hartree) -1.008678 :: r (angstrom) 1.437
* Energy (hartree) -1.002347 :: r (angstrom) 1.474
* Energy (hartree) -0.996374 :: r (angstrom) 1.511
* Energy (hartree) -0.990763 :: r (angstrom) 1.548
* Energy (hartree) -0.985517 :: r (angstrom) 1.585
* Energy (hartree) -0.980631 :: r (angstrom) 1.622
* Energy (hartree) -0.976101 :: r (angstrom) 1.659
* Energy (hartree) -0.971916 :: r (angstrom) 1.695
* Energy (hartree) -0.968065 :: r (angstrom) 1.732
* Energy (hartree) -0.964534 :: r (angstrom) 1.769
* Energy (hartree) -0.961307 :: r (angstrom) 1.806
* Energy (hartree) -0.958367 :: r (angstrom) 1.843
* Energy (hartree) -0.955696 :: r (angstrom) 1.880
* Energy (hartree) -0.953277 :: r (angstrom) 1.917
* Energy (hartree) -0.951092 :: r (angstrom) 1.953
* Energy (hartree) -0.949123 :: r (angstrom) 1.990
* Energy (hartree) -0.947353 :: r (angstrom) 2.027
* Energy (hartree) -0.945764 :: r (angstrom) 2.064
* Energy (hartree) -0.944341 :: r (angstrom) 2.101
* Energy (hartree) -0.943069 :: r (angstrom) 2.138
* Energy (hartree) -0.941933 :: r (angstrom) 2.175
* Energy (hartree) -0.940921 :: r (angstrom) 2.211
* Energy (hartree) -0.940019 :: r (angstrom) 2.248
* Energy (hartree) -0.939218 :: r (angstrom) 2.285
* Energy (hartree) -0.938506 :: r (angstrom) 2.322
* Energy (hartree) -0.937875 :: r (angstrom) 2.359
* Energy (hartree) -0.937315 :: r (angstrom) 2.396
* Energy (hartree) -0.936819 :: r (angstrom) 2.433
* Energy (hartree) -0.936380 :: r (angstrom) 2.470
* Energy (hartree) -0.935991 :: r (angstrom) 2.506
* Energy (hartree) -0.935648 :: r (angstrom) 2.543
* Energy (hartree) -0.935345 :: r (angstrom) 2.580
* Energy (hartree) -0.935077 :: r (angstrom) 2.617
* Energy (hartree) -0.934841 :: r (angstrom) 2.654
* Energy (hartree) -0.934633 :: r (angstrom) 2.691
* Energy (hartree) -0.934450 :: r (angstrom) 2.728
* Energy (hartree) -0.934289 :: r (angstrom) 2.764
* Energy (hartree) -0.934147 :: r (angstrom) 2.801
* Energy (hartree) -0.934022 :: r (angstrom) 2.838
* Energy (hartree) -0.933912 :: r (angstrom) 2.875
* Energy (hartree) -0.933816 :: r (angstrom) 2.912
* Energy (hartree) -0.933732 :: r (angstrom) 2.949
* Energy (hartree) -0.933658 :: r (angstrom) 2.986
* Energy (hartree) -0.933594 :: r (angstrom) 3.022
* Energy (hartree) -0.933537 :: r (angstrom) 3.059
* Energy (hartree) -0.933488 :: r (angstrom) 3.096
* Energy (hartree) -0.933445 :: r (angstrom) 3.133
* Energy (hartree) -0.933407 :: r (angstrom) 3.170
* Energy (hartree) -0.933374 :: r (angstrom) 3.207
* Energy (hartree) -0.933346 :: r (angstrom) 3.244
* Energy (hartree) -0.933321 :: r (angstrom) 3.280
* Energy (hartree) -0.933300 :: r (angstrom) 3.317
* Energy (hartree) -0.933281 :: r (angstrom) 3.354
* Energy (hartree) -0.933265 :: r (angstrom) 3.391
* Energy (hartree) -0.933251 :: r (angstrom) 3.428
* Energy (hartree) -0.933239 :: r (angstrom) 3.465
* Energy (hartree) -0.933228 :: r (angstrom) 3.502
* Energy (hartree) -0.933219 :: r (angstrom) 3.538
* Energy (hartree) -0.933211 :: r (angstrom) 3.575
* Energy (hartree) -0.933204 :: r (angstrom) 3.612
* Energy (hartree) -0.933199 :: r (angstrom) 3.649
Plot PECs
[12]:
import numpy as np
from scipy.interpolate import splrep, splev
import matplotlib.pyplot as plt
# step 1: read the data from the file
data_pyscf = np.loadtxt('pyscf_fci_sto3g_h2.data') #, skiprows=1) # Assuming the first row is a header
data_ibmq = np.loadtxt('h2_pec_ibmq_qasm_simulator.data')
# extract the columns
r = data_pyscf[:, 0]
energy = data_pyscf[:, 2]
r_ibmq = data_ibmq[:, 0]
energy_ibmq = data_ibmq[:, 1]
# standard deviation
std_ibmq = np.sqrt((data_ibmq[:, 2]))
# step 2: perform spline interpolation using SciPy
spline = splrep(r, energy, s=0) # Adjust s for smoothing factor
# Evaluate the spline at more points for a smoother curve
r_interp = np.linspace(r.min(), r.max(), 100)
energy_interp = splev(r_interp, spline)
# step 3: plot the interpolated data using Matplotlib
plt.figure()
plt.plot(r_interp, energy_interp, label='FCI/sto-3g', linewidth=2.5, color='gray')
plt.errorbar(r_ibmq, energy_ibmq, std_ibmq, label='qc2 + ibmq_qasm_simulator',
barsabove=False, ecolor='black', elinewidth=1.5,
linestyle='None', marker='s', mfc='white',
mec='black', ms=10*0.75, mew=2*0.75)
plt.xlabel('Bond length (angstrom)')
plt.ylabel('Energy (hartree)')
plt.title('Potential energy curve of H$_2$')
plt.legend()
plt.grid(False)
plt.show()